
Ubiquity is the new experimental extension to Firefox that will (I’m sure it will) make enormous impact on the way we use the browser. It allows to remix various services and extend functionality of the browser in very easy way (if you don’t get the point of Ubiquity yet, I recommend watching the video that came with official announcement; I needed to see that – description didn’t tell me much about how powerful it can be).
I didn’t have much time to play with it yet, but in spare 20 minutes I attempted to code a command that would show me the image of a structure from PDB given its code and eventually take me to its homepage. Suprisingly it was very easy (and I’m not a JS coder). The source is pasted below.
CmdUtils.CreateCommand({
name: "pdb",
description: "Goes to Protein Data Bank given PDB code.",
icon: "http://www.rcsb.org/favicon.ico",
help: "You can specify the PDB code and pressing enter will take you to particular structure's homepage." +
" If you type pdb code and press arrow down, you should see an image from PDB site.",
takes: {"PDB code": noun_arb_text},
execute: function( directObj) {
var pdbcode = directObj.text;
Utils.openUrlInBrowser("http://www.rcsb.org/pdb/explore/explore.do?structureId="+pdbcode);
},
preview: function( pblock, directObj ) {
var pdbcode = directObj.text;
pblock.innerHTML = "Preview of the structure:<br/>";
pblock.innerHTML += "<img src=\"http://www.rcsb.org/pdb/images/" + pdbcode + "_bio_r_250.jpg\" />";
}
})
It of course could be improved by using also a selected text, or allowing to keyword search the PDB (or basically any other biological database), but its current functionality suits me just fine. Ubiquity is not yet such a stable platform as Greasemonkey (or Chickenfoot), but it’s worth to keep an eye on it. I’m sure we will read sooner or later an article in peer-reviewed journal describing Ubiquity commands for life sciences :).
Related articles by Zemanta
![Reblog this post [with Zemanta]](https://i0.wp.com/img.zemanta.com/reblog_e.png)
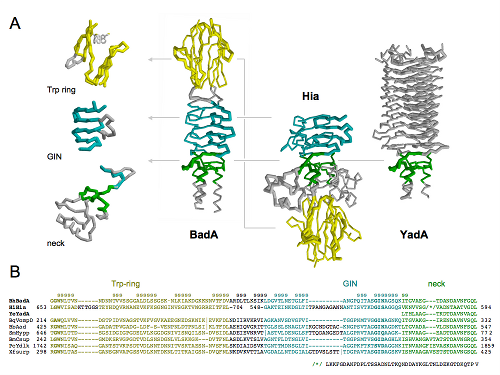
 Software for visualization of molecules is in majority of cases very focused on its job and rarely allows for something outside its scope (one of exceptions is
Software for visualization of molecules is in majority of cases very focused on its job and rarely allows for something outside its scope (one of exceptions is 




")





{kind=link}
How far one can push online collaboration in research?
A week or so ago I’ve asked on the FriendFeed if there’s an interest in writing a cyclic publication on the status of Science 2.0. I thought that summarizing every year advancements in openness of science would be a good idea. During discussion it turned out that there’s a need to write a first major publication on Science 2.0 concepts because there isn’t one published in the life sciences field. The final conclusion of interested people was to meet face to face on the upcoming conference and discuss things in detail.
And this made me think.
I started to wonder why people who live, breathe and do research online still need to meet in person to plan and discuss some stuff. The very obvious explanation is that the conference was only two weeks ahead, so there was nothing more than that, but some patterns (brainstorm online, then meet in person, then finish online) repeat so often that I started to believe an online collaboration can only go to a certain point.
The excuse is not in the tools, especially given how fast the new ones appear. As an examples may serve recently launched Adobe Acrobat site, which contains online editor and live collaboration suite (contains screen sharing, notes, chat, audio, video) or its analog for programmers: Assembla (svn, git, trac, wiki, milestones).
My feeling is that what makes a difference is not a quality of interaction while working on a certain project, but the possibility of discussing things not directly related to this project. Online collaboration is usually very focused. When editing a document online it’s usually hard to side-track it, so at the end it’s about something else than it was planned. Calls or videoconferences have usually a schedule. No place for non-related stuff. No place for a beer/coffee/glass of water and a chat about how life was good in old times. No place for discussions on random things and coming to the main project from a different angle and with completely new ideas.
I’ve been trying for quite a long to work online with other people on some projects from my home office (which is in the middle of nowhere). As you can guess, it works to a certain point and majority of them were improved upon meeting face to face. And this left me wondering how far can I push online collaborations. It isn’t usually an issue in IT field, but so far it doesn’t look very promising in research.
Posted by Pawel Szczesny on August 27, 2008 in Comments, Community, Research
Tags: FriendFeed, Science 2.0