With my background in protein sequence analysis and recently increasing interest in synthetic biology/biological engineering, Deepak’s post about BioBrick project immediately caught my eye. However, after spending some time around registry of standard biological parts I realized that using full natural proteins as modules wasn’t very appealing to me. My thoughts oscillated in area between nanotechnology at a atomic level, and BioBrick approach – my idea for “a brick” was something from supersecondary structure elements to protein domains.
While I was trying to figure out what was done in this area, here’s what appeared in Google Reader: Peptide and Protein Building Blocks for Synthetic Biology: From Programming Biomolecules to Self-Organized Biomolecular Systems by E.H.C. Broomley, K. Channon, E. Moutevelis and D.N. Woolfson (DOI: http://dx.doi.org/10.1021/cb700249v). It’s not an open access paper, but you can download it from Dek Woolfson lab page. This review summarizes several different approaches to synthetic biology focusing specifically on peptides and proteins as a prime component of newly engineered machines. Why actually peptides and proteins? Here are some points from the paper:
- efficient, reproducible and spontaneous folding with all information usually encoded in the polypeptide chain
- organization of the folded structures, including secondary structure elements (referred there as tectons), which naturally limits the possible number of folds, but gives enough different structural scaffolds to display many biological functions
- frequently observed self-assembly of folded structures into higher-order complexes/nanomachines
The review gives examples of selected protein folding motifs (like collagens or zinc-fingers) that can be used in designing novel fibrous materials, but describes in great detail various assemblies made of protein coiled-coils.
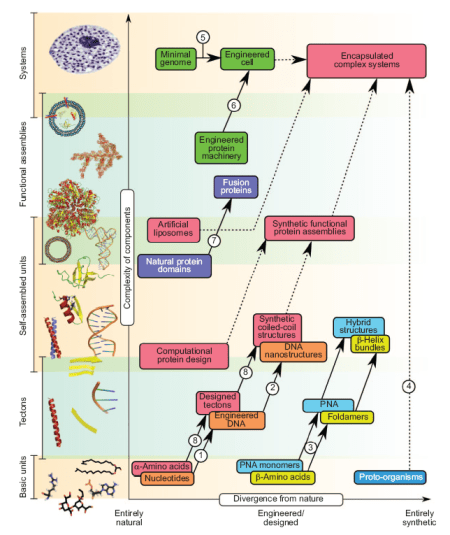
Being pretty familiar with coiled-coil structures, I was rather more interested in general view on the field – and I wasn’t disappointed. Below I copied first figure from this paper (for its legend see the original paper) – it’s a beautiful breakdown of different methodologies in synthetic biology. You can place on this graph most approaches people are taking in this area (from Venter’s bacteria, through BioBricks, to DNA cubes).


![Reblog this post [with Zemanta]](https://i0.wp.com/img.zemanta.com/reblog_e.png)











Thoughts on CASP – Critical assessment of methods of protein structure prediction
I’ve just read an introduction to the supplemental issue of the journal PROTEINS, dedicated to the most recent round of the CASP experiment. It describes the progress of the protein structure prediction over the last few CASP editions.
The list of advancements include:
I believe that this was possible thanks to the progress that has been made in the area of sequence homology searches. Finding similarity between two sequences well beyond any reasonable identity thresholds is now doable thanks to profile-to-profile comparison, meta-servers (joining predictions from many different methods) or recent hmm-to-hmm algorithms (comparison of Hidden Markov Models). If you can find a suitable template for your protein, the rest is then much easier, isn’t it?
There are of course fields that still need some work. One of these often stirs a lot of discussion: automated assessing of model similarity to the real structure. The current methods have proven their suitability, I definitely agree. However I hope that at some point the protein structure comparison software will refuse to superimpose eight- and ten-stranded beta-barrels or left- and right-handed coiled-coil with a message: “It doesn’t make sense.”
Posted by Pawel Szczesny on October 10, 2007 in Comments, Papers, Research, Structure prediction
Tags: bioinformatics, casp, Proteins, Research, Structure prediction