This post starts a series devoted to imaginary nanodevices made of proteins. I’m going to play around with known protein structures to see if some of them can form an interesting arrangement. Basic requirement is lack of obvious sterical clashes at the level of a main chain trace. If that is fulfilled I would assume very slight chance that particular arrangement is possible. However, in most cases I won’t bother inventing how to recreate it in the lab, since I don’t feel competent enough. The whole series is more fiction than science and my goal is mainly stretching my and readers imagination.
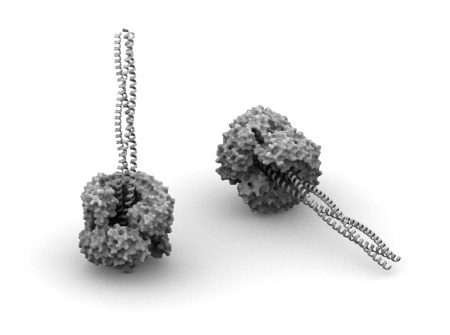
Lets start with something simple. Structure depicted above is a dimer of leucin rich repeat (LRR) protein (PDB: 1A4Y, chains A and D) with a trimeric coiled-coil (my own model made with BeammotifCC) fitted in. The opening is wide enough to accommodate three helices without any problems. Picture below shows main chain trace of the coiled-coil (in red) surrounded by LRR dimer (all atoms, blue and sea green). As you can see, any coiled-coil made of aminoacids with small side chains would not create any sterical issues. In fact, approximate size of the opening (~35 Angstroms) is much larger than the opening size of the membrane anchor of trimeric autotransporter adhesins (twelve stranded beta-barrel, PDB: 2GR7), which also accommodates a trimeric coiled-coil. So why not to use a beta-barrel instead of LRR? Well, beta-barrels are hardly present outside membranes 🙂 .

One can ask question if the single LRR protein can make a full ring. It looks possible from the structure of the single repeat (beta-turn-alpha) – interactions with preceding and following repeats are virtually the same. However, secondary structure elements of these repeats are not perfectly aligned with the axis of the opening. Their tilt forces consecutive repeats to form an imaginary spiral, not a circle (although the tilt does not seem to be large enough to actually allow for spiral folding of larger number of repeats – but that’s only my assumption, it would be worth to check).
So that’s it for now. If you feel that I’m rediscovering wheel, writing something completely silly, or you have any suggestions, please feel free to discourage/encourage me with comments.







Thoughts on CASP – Critical assessment of methods of protein structure prediction
I’ve just read an introduction to the supplemental issue of the journal PROTEINS, dedicated to the most recent round of the CASP experiment. It describes the progress of the protein structure prediction over the last few CASP editions.
The list of advancements include:
I believe that this was possible thanks to the progress that has been made in the area of sequence homology searches. Finding similarity between two sequences well beyond any reasonable identity thresholds is now doable thanks to profile-to-profile comparison, meta-servers (joining predictions from many different methods) or recent hmm-to-hmm algorithms (comparison of Hidden Markov Models). If you can find a suitable template for your protein, the rest is then much easier, isn’t it?
There are of course fields that still need some work. One of these often stirs a lot of discussion: automated assessing of model similarity to the real structure. The current methods have proven their suitability, I definitely agree. However I hope that at some point the protein structure comparison software will refuse to superimpose eight- and ten-stranded beta-barrels or left- and right-handed coiled-coil with a message: “It doesn’t make sense.”
Posted by Pawel Szczesny on October 10, 2007 in Comments, Papers, Research, Structure prediction
Tags: bioinformatics, casp, Proteins, Research, Structure prediction