First major outcome of my PhD project has just appeared in the Bioinformatics (open access). It describes a system we have design to annotate specific group of bacterial proteins.
Trimeric autotransporter adhesins (TAAs) form one of the many families of bacterial surface proteins. In medically relevant species they adhere to host cells (in non-pathogenic species we don’t know what they adhere to), therefore they are considered essential virulence factors. They are autotransporters, which means that they are passing the outer membrane by themselves – C-terminal part makes a pore through which the rest of the protein goes out. In contrary to many other autotransporters, exported part is not cut but stays attached to the membrane by the C-terminal autotransport domain. TAAs are also trimeric – the pore is made of three subunits and the exported fiber is also a trimer. The last feature is pretty unique – so far it’s the only family of bacterial surface proteins which forms fibrous trimers. Interestingly, they differ in length between few hundred and five thousands residues.
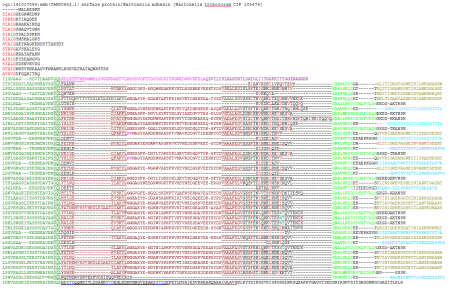
What’s so special about these proteins for bioinformatician? Structure of the fiber is not homogenous – it is a mixture of globular domains and coiled-coils. On a sequence level, they have lots of internal repeats (see the picture), heavily biased residue composition, their domain composition and architecture varies by protein. The only conserved part in all TAAs in the autotransport domain. Systems designed to identify and annotate typical protein domains (such as PFAM) don’t handle them very well – average coverage of PFAM annotation of TAAs is about 30%. The server we have built relies on the fact that domains of TAAs are exclusive for this family (they do not appear anywhere else because its unique structural constrains). Therefore we could use different thresholds, manually curated alignments and domain-context derived rules to improve the annotation.
Manual analysis of TAAs sequences is pretty tedious (well, it was, now we have the server), but on the other hand I have learnt a lot about how to read a protein sequence. I mean really read and understand how particular combination of letters influences its structure.







ludo
April 10, 2008 at 21:05
Congratulation pawel, thats a good news, and bioinformatics is a nice journal!
did you have any structural information? xray or modeling? Is it the trimer that bind the membrane and pass through it ?
Maybe you should be interested on some docking technics that can permit to explore symetrical assembly….For my point of view MZ-Dock is the best ones.
Thus if there is no xray of the trimer or the monomer, maybe it will be interesting to modelize it and explore the different possible trimer…
Congratulation again
Pawel Szczesny
April 11, 2008 at 16:57
Thanks Ludovic. There are a few crystal structures for small parts of this family and for the autotransport domain. Exported parts don’t really resemble typical proteins – for example significant portion of the trimeric fiber is heavily interwound so a monomer doesn’t make sense without other two subunits (see for example 1s7m). We have no idea how the autotransport works, but current assumption is that all three chains go through the pore unfolded and assemble outside of the cell.
So docking of individual monomers is not really necessary, as modelling (when possible) already includes all three subunits (otherwise structure of the monomer collapses in minimization). Thanks for the software suggestion, but I cannot find MZ-Dock anywhere – can you post a link?
ludo
April 12, 2008 at 07:03
Yes your right its seems that docking will bring no furhter information, but perhaps you can get information normal mode analysis (elesticity and deformation of the trimer fiber). Anyway, if you need to get a trimer, or dimer symetrical you should use http://zlab.bu.edu/m-zdock/ (the paper here http://bioinformatics.oxfordjournals.org/cgi/content/full/21/8/1472). I have test all symetrical docking software, it is the simplest and most efficiant one.
Is there a possibility that your protein act like a porine?